Sterility Testing for Research Peptides: A Protocol Guide

Sterility testing is defined as the microbiological assay that confirms the absence of viable bacteria, fungi, and other microorganisms in a peptide sample, making it a biological quality attribute entirely separate from chemical purity or potency. The role of sterility testing in research peptides is to catch contamination that HPLC chromatograms and mass spectrometry cannot detect. USP <71> is the regulatory backbone governing this assay for injectable and research-grade materials. Sterility testing also works alongside endotoxin testing and purity analysis to form a complete quality control picture. Without all three, a researcher cannot accurately assess whether a peptide batch is safe and reliable for use.
What are the standard sterility testing methods for research peptides?
USP <71> defines two compendial methods for sterility testing: membrane filtration and direct inoculation. Both are harmonized with European Pharmacopoeia (EP) and Japanese Pharmacopoeia (JP) standards, which means a test performed under one framework is generally recognized across all three.
Membrane filtration is the preferred method for soluble peptides. The sample passes through a 0.45-micron membrane that retains microorganisms. The membrane is then transferred to culture media and incubated. This approach removes antimicrobial components from the sample before incubation, which reduces interference and improves detection sensitivity.

Direct inoculation is used when membrane filtration is not feasible, typically for viscous or insoluble formulations. The sample is added directly to culture media without filtration. This method carries a higher risk of false negatives when the peptide itself has antimicrobial properties.
Both methods use two culture media types:
Thioglycollate medium: Incubated at 30–35°C for 14 days; detects aerobic and anaerobic bacteria.
Soybean-casein digest medium (SCDA): Incubated at 20–25°C for 14 days; detects fungi and aerobic bacteria.
The table below summarizes key protocol parameters under USP <71>:
Parameter Thioglycollate medium Soybean-casein digest medium Temperature 30–35°C 20–25°C Incubation period 14 days 14 days Target organisms Anaerobes, aerobes Fungi, aerobes Minimum sample units ≥20 per batch ≥20 per batch Acceptance criterion Zero growth Zero growth
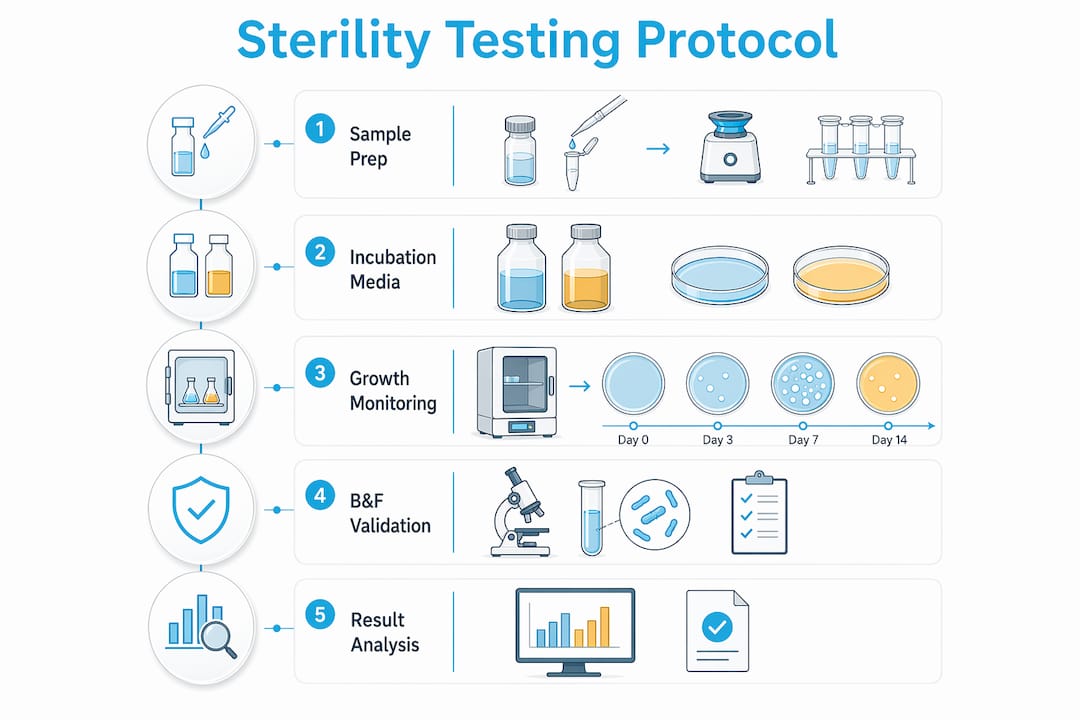
Sterility testing is qualitative presence/absence testing. A single contaminated unit triggers batch rejection, which reflects a zero-tolerance standard for viable microorganisms in released product. Rapid Microbial Methods (RMM) such as ATP bioluminescence and DNA microarrays can reduce turnaround to 24–48 hours, but these techniques serve as early screening tools only. The 14-day traditional culture method remains mandatory for final batch release under USP <71>.
Why is method suitability validation critical for peptide sterility testing?
Method suitability validation, formally called bacteriostasis and fungistasis (B&F) testing, confirms that the peptide sample does not suppress microbial growth in the culture media. This step is mandatory under USP <71> and is the most commonly overlooked element in research peptide sterility testing protocols.
Many peptides carry inherent antimicrobial activity. Cationic peptides, for example, disrupt bacterial membranes. If such a peptide inhibits growth in the test media, a contaminated sample can appear sterile. The result is a false negative, which is arguably more dangerous than a failed test.
The B&F procedure follows a defined sequence:
Select six USP-specified microorganisms: Staphylococcus aureus, Bacillus subtilis, Pseudomonas aeruginosa, Clostridium sporogenes, Candida albicans, and Aspergillus brasiliensis.
Inoculate each organism at ≤100 CFU into the test media containing the peptide sample.
Inoculate a control set of the same media without the peptide sample.
Compare growth between the two sets after incubation.
Confirm that growth in the peptide-containing media is equivalent to the control.
If the peptide inhibits growth in any organism, the method fails B&F validation. The lab must then modify the approach, typically by increasing the dilution factor, adding neutralizing agents such as polysorbate 80 or lecithin, or switching to membrane filtration to physically remove the antimicrobial components before inoculation.
False negatives from antimicrobial interference make B&F testing mandatory, not optional. Regulatory agencies treat a sterility test performed without valid B&F data as an incomplete assay. Researchers sourcing peptides should verify that the COA explicitly states method suitability was confirmed before accepting sterility test results.
Pro Tip: Ask your supplier for the B&F validation report, not just the sterility test result. If they cannot provide it, the sterility data on the COA is not reliable.
What are the limitations and misconceptions about sterility testing?
Sterility testing does not prove absolute sterility. This is the most consequential misconception in peptide quality control, and it affects how researchers should interpret COA data.
The assay is a statistical sampling procedure. A batch of 1,000 vials might have 10 contaminated units. If the 20 units sampled for testing happen to be uncontaminated, the batch passes. The sterility assurance level (SAL) achieved by testing alone reaches only approximately 10⁻², far short of the 10⁻⁶ SAL required for injectable products. That 10⁻⁶ level is achieved through validated sterilization processes and ISO Class 5 clean room environments, not through the test itself.
Additional limitations include:
Viable but not culturable (VBNC) organisms: Some bacteria enter a dormant state and do not grow in standard culture media, making them invisible to the assay.
Slow-growing organisms: A 14-day incubation may not be sufficient for all fungal species or atypical bacteria.
Laboratory contamination: False positives can occur when the testing environment itself introduces organisms, invalidating the result.
Assay interference: As discussed in the B&F section, antimicrobial peptide components can suppress growth and generate false negatives.
Passing a sterility test confirms that no contamination was detected within the limits of the sampling and culture conditions used. It does not confirm that the entire batch is free of viable microorganisms. Researchers who treat a passed sterility test as proof of absolute sterility are misapplying the data.
The distinction between sterility and purity also requires clarity. A peptide with 99% HPLC purity can still harbor viable microorganisms. Purity measures chemical composition. Sterility measures biological contamination. These are independent quality attributes with no overlap, and neither result informs the other.
How does sterility testing complement endotoxin and purity testing?
Sterility, endotoxin, and purity testing address three distinct quality questions. No single assay substitutes for the others, and assuming otherwise creates real risk in research applications.
HPLC purity testing measures the chemical composition of a peptide. A result of 98% purity means 98% of the detected material is the target peptide. It says nothing about whether the sample contains live bacteria, fungal spores, or bacterial cell wall fragments. High HPLC purity does not correlate with sterility or endotoxin levels.
Endotoxin testing, typically performed using the Limulus Amebocyte Lysate (LAL) assay, detects lipopolysaccharide (LPS) from gram-negative bacterial cell walls. LPS survives sterilization. A sample can pass sterility testing because all organisms are dead, yet still fail endotoxin testing because LPS persists after sterilization. This is a critical distinction for any peptide intended for injection or cell culture use.
Test What it detects Method Limitation Sterility (USP <71>) Viable microorganisms Culture-based Statistical sampling; misses VBNC organisms Endotoxin (LAL assay) Bacterial LPS residues Enzymatic reaction Does not detect live organisms or purity issues Purity (HPLC) Chemical composition Chromatography Does not detect biological contamination
Sterility and endotoxin testing are not interchangeable. A COA that lists only HPLC purity is missing two of the three critical quality dimensions. Researchers working with peptides in cell-based assays, animal models, or any injectable application need all three data points to make an informed assessment of batch quality.
Pro Tip: When reviewing a COA, treat a missing sterility or endotoxin line as a gap in the data, not as a passed result. Absence of the test is not reassurance.
What practical steps should researchers take with sterility test data?
Interpreting sterility test results correctly requires understanding both what the data confirms and what it does not. The reliability of sterility results depends directly on whether an ISO 17025 accredited laboratory performed the assay with validated method suitability for the specific peptide matrix.
When reviewing a COA for sterility data, check for the following:
Lab accreditation: The testing laboratory should hold ISO 17025 accreditation. This confirms the lab operates under a quality management system with validated methods.
Method suitability statement: The COA or accompanying documentation should confirm B&F validation was performed and passed.
Both sterility and endotoxin results: A COA that lists sterility but omits endotoxin data, or vice versa, is incomplete for most research applications.
Batch-specific data: Sterility results should reference the specific batch number, not a generic product-level claim.
Pass/fail interpretation: A pass means no growth was detected in the sampled units under the stated conditions. It does not mean the entire batch is sterile.
COAs frequently omit sterility and endotoxin data, and researchers should treat that absence as a signal that the tests were not performed. Asking a supplier directly whether sterility testing was conducted, and requesting the underlying lab report, is a reasonable and necessary step in vendor qualification. Guidance on evaluating supplier credentials can help researchers structure those conversations.
Aseptic handling after receipt is equally important. A peptide that passes sterility testing at the point of release can become contaminated during reconstitution if the researcher uses non-sterile water for injection, unfiltered solvents, or non-sterile equipment. Sterility testing covers the product at release. Post-release contamination is the researcher’s responsibility.
Pro Tip: Reconstitute lyophilized peptides using bacteriostatic water or sterile water for injection, and use a 0.22-micron syringe filter when preparing solutions for sensitive applications. This preserves the sterility status of the released product.
Understanding peptide safety testing in a broader context helps researchers connect sterility data to the overall risk profile of a compound, particularly for injectable applications.
Key Takeaways
Sterility testing confirms the absence of viable microorganisms in a peptide batch, but it requires validated method suitability, complementary endotoxin testing, and correct COA interpretation to deliver reliable quality assurance.
Point Details USP <71> governs sterility testing Two methods apply: membrane filtration and direct inoculation, both with 14-day incubation periods. B&F validation is mandatory Method suitability testing must confirm the peptide does not suppress microbial growth in culture media. Sterility testing has statistical limits A passed test confirms no contamination detected in sampled units, not absolute sterility of the batch. Three tests cover three quality domains Purity, sterility, and endotoxin testing answer different questions and none substitutes for the others. COA gaps signal missing tests Absent sterility or endotoxin lines on a COA typically mean the test was not performed, not that it passed.
The test result is only as good as the method behind it
Researchers often focus on the pass/fail outcome on a COA and overlook the conditions that produced it. After reviewing sterility documentation across a range of research peptide suppliers, the pattern is consistent: sterility test results appear on COAs without any reference to B&F validation, lab accreditation, or the specific method used. A pass result generated without valid method suitability data is not a reliable result. It is a number without context.
The more useful question is not “did this batch pass sterility testing?” but “was the sterility test performed under conditions that would actually detect contamination?” Those are different questions, and the second one is rarely asked.
Endotoxin testing deserves equal attention. Researchers working in cell culture frequently attribute unexpected inflammatory responses to the peptide itself, when the actual cause is LPS contamination in a batch that passed sterility testing. The organisms were dead. The LPS was not. A comprehensive quality assessment that includes sterility, endotoxin, and purity data would have flagged the issue before the experiment ran.
The realistic expectation for sterility testing is this: it is a necessary but insufficient quality layer. It catches gross contamination events in sampled units. It does not guarantee the entire batch is clean, and it does not detect chemical impurities or endotoxin residues. Researchers who understand these boundaries use the data correctly. Those who do not tend to over-rely on a single test result and under-invest in the other quality parameters that matter equally.
— Sam Levin
Research-grade peptides with documented sterility and endotoxin testing
PeptidesFromChina supplies research peptides accompanied by batch-specific Certificates of Analysis that include sterility and endotoxin testing data alongside HPLC purity results. The platform works directly with synthesis facilities that conduct USP <71> sterility testing and LAL-based endotoxin assays, with documentation available for researcher review before purchase.
Researchers who need peptides with complete biological and chemical quality documentation can review the full peptide catalog or examine specific products such as research-grade KPV, which includes sterility, endotoxin, and purity data on the accompanying COA. PeptidesFromChina prioritizes batch traceability and supplier transparency over volume-based reseller models, making it a practical sourcing option for laboratories that require documented quality control across all three testing domains.
FAQ
What does USP <71> require for peptide sterility testing?
USP <71> requires incubation in thioglycollate medium at 30–35°C and soybean-casein digest medium at 20–25°C, both for 14 days, with a minimum of 20 units sampled per batch. Any microbial growth results in batch rejection.
What is bacteriostasis and fungistasis testing?
B&F testing confirms that the peptide sample does not inhibit microbial growth in the sterility test media. It uses six USP-specified organisms inoculated at ≤100 CFU each, and failure requires method modification before sterility results are considered valid.
Can a peptide pass sterility testing and still contain endotoxins?
Yes. Endotoxins are LPS fragments from bacterial cell walls that survive sterilization. A batch can pass sterility testing because all organisms are dead, yet still carry endotoxin levels that cause inflammatory responses in cell or animal models.
Does high HPLC purity mean a peptide is sterile?
No. Purity and sterility are independent quality attributes. A peptide with 99% HPLC purity can still contain viable microorganisms or endotoxin residues that purity testing cannot detect.
How should researchers interpret a missing sterility result on a COA?
Absent sterility or endotoxin data on a COA typically indicates the test was not performed, not that the result was satisfactory. Researchers should request the underlying lab report or ask the supplier directly whether testing was conducted.