Peptide Synthesis Partner Criteria: A Researcher’s Guide

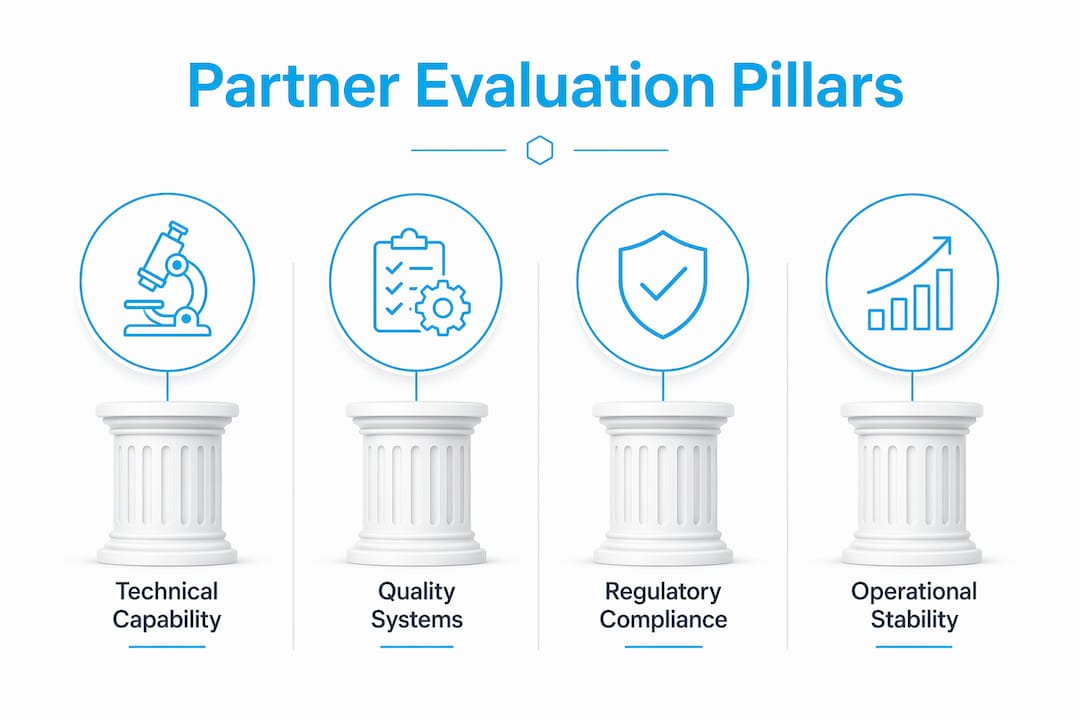
Selecting a peptide synthesis partner is defined by four core evaluation pillars: technical capability, quality systems maturity, regulatory compliance, and operational stability. Each pillar carries a distinct weight in the decision, with technical capability at 25–30% and quality systems at 20–25% of the overall assessment. Researchers who skip a structured evaluation framework often end up with inconsistent batches, missing documentation, or suppliers who cannot scale when projects advance. The select peptide synthesis partner criteria covered here reflect real manufacturing realities, not marketing claims, and apply whether you are sourcing milligram quantities for in vitro work or planning gram-scale in vivo studies.
What are the core criteria for selecting a peptide synthesis partner?
Peptide synthesis partner selection begins with matching technical depth to project requirements. A supplier capable of producing a simple 15-residue catalog peptide is not automatically equipped to handle a 60-residue cyclic peptide with unnatural amino acids. The gap between those two tasks is significant, and conflating them is one of the most common sourcing errors researchers make.

Generic vendor checklists designed for pharmaceutical outsourcing often miss peptide-specific technical requirements entirely. Solid-phase peptide synthesis capacity, purification expertise, and analytical instrumentation are not standard line items in most procurement templates. Researchers need a peptide-specific framework that weights these factors correctly from the start.
A structured vendor evaluation that predefines scoring criteria before engaging suppliers removes bias and produces more defensible decisions. Weighting technical capability, quality systems, regulatory compliance, and financial stability in advance forces the evaluation to stay objective. Without predefined weights, procurement decisions tend to drift toward price or familiarity rather than fit.
What technical capabilities should a synthesis partner have?
Synthesis method and sequence complexity
Solid-phase peptide synthesis using Fmoc or Boc chemistry is the industry standard for research-grade peptide production. Fmoc is more common for research applications due to milder deprotection conditions. The synthesis method itself matters less than the supplier’s demonstrated experience with the sequence complexity your project requires.
Standard synthesis handles peptides in the 30–50 amino acid range reliably. Sequences beyond 100 residues, or those incorporating cyclic structures, PEGylation, phosphorylation, or non-natural amino acids, require specialized facilities and process development experience. Asking a supplier for examples of comparable sequences they have successfully produced is a direct way to assess this.
Routine synthesis: linear peptides, 30–50 residues, standard amino acids
Intermediate complexity: disulfide bridges, isotopic labeling, C-terminal amidation
High complexity: cyclic peptides, PEGylated conjugates, 100+ residues, unnatural amino acids
Purification method: preparative reverse-phase HPLC is the standard; confirm column chemistry and gradient protocols
Analytical instrumentation and in-house QC
Purification alone does not confirm identity or purity. In-house analytical capabilities, specifically HPLC and mass spectrometry, determine whether a supplier can generate batch-specific data rather than relying on shared or outsourced testing. Outsourced analytical work introduces delays and reduces traceability.
Pro Tip: Ask whether the HPLC and mass spec instruments used for QC are on-site. If a supplier sends samples to a third-party lab for routine analysis, batch turnaround times and data integrity both become harder to verify.
A supplier’s SPPS capacity and purification setup should be confirmed through facility documentation, not just a sales conversation. Resin loading capacity, synthesizer scale, and HPLC column dimensions all indicate whether a supplier can realistically handle your batch size and timeline.
How do quality control and batch documentation affect peptide reliability?
Batch-specific documentation is the single most reliable indicator of a supplier’s quality commitment. A Certificate of Analysis that includes an actual HPLC chromatogram, mass spectrometry confirmation, and a net peptide content statement reflects genuine QC practice. A COA that lists only a purity percentage with no supporting data is a risk indicator, not a quality assurance document.
The distinction between gross weight and net peptide content is frequently misunderstood and routinely exploited. Net peptide content differs from gross weight because lyophilized peptides retain moisture and counterions that add mass without adding active compound. A peptide reported at 95% purity by HPLC may contain only 70–80% actual peptide by weight when moisture and salt content are factored in. Dosing calculations based on gross weight rather than net content introduce systematic errors into every experiment.
Key documentation standards to require from any supplier:
Batch-specific HPLC chromatogram with integration data and method parameters
Mass spectrometry confirmation of molecular weight (ESI-MS or MALDI-TOF)
Net peptide content assay, not just gross weight purity
Solubility testing results where relevant
Residual solvent and counterion data for GLP-level work
Pro Tip: A high purity percentage means nothing if the HPLC method is poorly validated. Review batch chromatograms directly, check peak symmetry, and confirm the integration baseline is set correctly. A single broad shoulder on a chromatogram can indicate a closely eluting impurity that a percentage alone will not reveal.
Quality systems maturity varies significantly across suppliers. ISO 9001 certification covers process management and is appropriate for discovery-stage research. GLP compliance is required for IND-enabling studies. GMP is necessary for clinical-grade material. Paying for GMP synthesis during early discovery is unnecessarily costly and does not improve data quality at that stage.
What regulatory and contractual safeguards are required?
Matching certification level to project stage is a practical and financial decision. The certification hierarchy works as follows:
ISO 9001: Covers quality management systems and process documentation. Appropriate for research-grade catalog peptides and early discovery work.
GLP (Good Laboratory Practice): Required for studies that will support regulatory submissions, including IND-enabling toxicology and pharmacokinetics work.
GMP (Good Manufacturing Practice): Required for clinical-grade material used in human trials. Involves facility qualification, validated processes, and extensive batch records.
Regulatory track record matters beyond certification status. A supplier with no history of FDA or EMA warning letters or observations demonstrates sustained compliance, not just a certificate on the wall. Requesting a supplier’s regulatory history is standard practice in pharmaceutical outsourcing and should be equally standard in peptide procurement.
Intellectual property protection is the most frequently neglected area in peptide synthesis contracting. The most costly mistake researchers make is disclosing peptide sequences before a signed Confidential Disclosure Agreement or Non-Disclosure Agreement is in place. Once a sequence is shared without contractual protection, enforcing exclusivity becomes legally difficult and practically impossible in many jurisdictions.
A complete contract for custom peptide synthesis should address:
CDA or NDA signed before any sequence or structural data is disclosed
Explicit IP ownership clauses confirming the researcher retains full rights
Prohibition on sequence reuse, resale, or disclosure by the supplier
Data destruction or return protocols upon project completion
Clear distinction between catalog research peptides and custom synthesis deliverables
Transparency in service scope and labeling, including Research Use Only designations on catalog products, reflects a supplier’s operational maturity. Suppliers who blur the line between RUO catalog peptides and clinical-grade material create compliance risk for the researcher, not just for themselves. A supplier qualification program that classifies vendors by risk level and includes documentation audits provides a structured way to assess this before committing to a supplier.
How does operational stability affect long-term peptide supply?
Supply scale requirements change as research advances, and a partner who cannot grow with the project creates timeline risk. Manufacturing capacity needs to match the project stage: milligrams for in vitro cell assays, grams for animal studies, and kilograms for late-stage or commercial manufacturing. A supplier who is transparent about their capacity ceiling is more reliable than one who overpromises and underdelivers.
The table below contrasts two common supplier categories researchers encounter:
Evaluation factor Specialized synthesis CRO Catalog peptide reseller Custom sequence capability Yes, with process development Limited or none Batch-specific COA Standard Variable; often generic Scale flexibility mg to kg with planning Typically fixed catalog quantities IP and NDA protocols Formal contracts standard Often absent Regulatory documentation GLP or GMP available Rarely available Supply chain transparency Direct facility access Often opaque; intermediary model
Resellers operating through intermediary channels introduce traceability gaps that are difficult to close after the fact. When a batch fails and the supplier cannot provide the original synthesis facility’s documentation, root cause analysis becomes impossible. Direct sourcing from API manufacturers rather than resellers reduces this risk substantially.
Financial and operational stability also affect supply reliability. A supplier with high staff turnover, inconsistent lead times, or vague answers about surge capacity is signaling operational fragility. Asking for references from researchers who have placed repeat orders over 12 or more months provides more useful information than any sales presentation.
Key Takeaways
Selecting a peptide synthesis partner requires matching technical capability, quality documentation, regulatory certification, and supply chain transparency to the specific demands of each project stage.
Point Details Match certification to project stage Use ISO 9001 for discovery, GLP for IND-enabling studies, and GMP only for clinical-grade material. Require batch-specific COA Demand HPLC chromatograms, mass spec data, and net peptide content, not just a purity percentage. Protect IP before disclosure Sign a CDA or NDA before sharing any peptide sequence with a prospective supplier. Verify net peptide content Dosing errors result from using gross weight; always calculate from net peptide content assay data. Assess supply chain transparency Prefer suppliers with direct facility access and traceable batch records over opaque reseller models.
What I’ve learned about evaluating synthesis partners that most guides skip
Most articles on choosing peptide synthesis providers stop at certification checklists. The real differentiators show up in how a supplier responds to specific technical questions, not in what their website claims.
The most underestimated risk is purity misinterpretation. Researchers routinely accept a stated purity figure without asking which HPLC method produced it, what column was used, or whether the integration was manually reviewed. A supplier who can answer those questions without hesitation is operating at a different level than one who redirects to a PDF. Insisting on raw chromatogram files, not just summary tables, is the fastest way to separate genuine QC from documentation theater.
Communication quality is also a reliable proxy for operational maturity. A supplier who responds to technical questions with specific, accurate answers, including honest acknowledgment of limitations, is demonstrating the same rigor they apply to synthesis. Vague or evasive responses to questions about net peptide content, counterion removal, or lyophilization conditions are meaningful signals.
The advice to avoid GMP synthesis in early discovery is worth repeating. Researchers sometimes pursue GMP-grade material for in vitro screening work because it feels more rigorous. It is not more rigorous for that application. It is more expensive and slower, and it does not improve the quality of discovery-stage data. Matching quality system requirements to actual project needs is a discipline, not a compromise.
A weighted scoring framework built before vendor engagement, with criteria defined by the project’s technical and regulatory requirements, consistently produces better sourcing decisions than intuition or price comparison alone.
— Sam Levin
PeptidesFromChina: research-grade peptides with transparent batch verification
PeptidesFromChina operates as a research-focused sourcing platform with direct relationships with established synthesis facilities, not as a gray-market reseller.
Every peptide in the research catalog ships with a batch-specific Certificate of Analysis that includes HPLC chromatogram data, mass spectrometry confirmation, and net peptide content. Custom synthesis requests follow the same documentation standards, with full batch traceability from raw API through lyophilization and vialing. For researchers who need verified research-grade peptides with supply chain transparency built in, PeptidesFromChina provides direct access to VIP-tier research peptides and catalog compounds backed by independent QC data.
FAQ
What are the four pillars of peptide synthesis partner evaluation?
The four core pillars are technical capability, quality systems maturity, regulatory compliance, and operational stability. Each carries a defined weight in a structured evaluation, with technical capability typically weighted at 25–30%.
What should a peptide Certificate of Analysis include?
A reliable COA includes a batch-specific HPLC chromatogram, mass spectrometry confirmation of molecular weight, and a net peptide content statement. A COA that lists only a purity percentage without supporting raw data is insufficient for research use.
When is GMP synthesis actually required?
GMP synthesis is required for peptides used in human clinical trials. For discovery-stage research and IND-enabling studies, ISO 9001 and GLP compliance, respectively, are the appropriate standards. GMP at the discovery stage adds cost without improving data quality.
How does net peptide content differ from gross weight?
Net peptide content accounts for moisture and counterions retained during lyophilization, which reduce the actual amount of active peptide relative to total mass. Dosing calculations must use net content to avoid systematic errors in experimental results.
What IP protections are required before sharing a peptide sequence?
A signed Confidential Disclosure Agreement or Non-Disclosure Agreement must be in place before any sequence data is disclosed to a supplier. The contract should explicitly state IP ownership, prohibit sequence reuse, and define data destruction or return protocols.