How to Assess Peptide Supplier GMP Compliance in 2026

Assessing peptide supplier GMP compliance is defined as the systematic verification of a supplier’s regulatory certifications, batch documentation, analytical testing records, and quality management practices against recognized pharmaceutical manufacturing standards. The formal industry term for this process is supplier qualification, and it encompasses everything from certificate authentication to ongoing performance monitoring. Regulatory bodies including the FDA, EMA, and NMPA set the baseline requirements, while tools like batch-specific Certificates of Analysis (CoA), HPLC purity data, and independent mass spectrometry testing provide the evidence that compliance is real rather than claimed. For QA and procurement teams, a structured approach to peptide supplier evaluation is the only reliable defense against batch failures, regulatory exposure, and supply chain disruption.
How to assess peptide supplier GMP compliance through documentation
The first step in any supplier qualification process is verifying that GMP certificates are genuine, current, and issued by a recognized regulatory authority. Self-issued GMP certificates and ISO certifications alone do not prove GMP compliance. ISO 9001 addresses quality management systems, not pharmaceutical manufacturing controls. A supplier claiming GMP status must produce a certificate traceable to the FDA, EMA, or China’s NMPA, and that certificate must name the specific manufacturing site.
Independent verification is non-negotiable. The FDA’s establishment registration database, the EudraGMP portal for European facilities, and the NMPA’s online license registry all allow procurement teams to confirm that a facility’s registration is active and matches the address on the supplier’s documentation. A certificate that cannot be cross-referenced in one of these official databases is not a valid GMP credential.

Beyond the certificate itself, the CoA is the most operationally critical document in any GMP compliance check. A proper CoA includes the peptide name and amino acid sequence, a unique batch number, the testing date, HPLC purity results with method details, mass spectrometry confirmation data, and acceptance criteria listed alongside actual results. Generic “Pass” statements without numeric purity values, missing batch numbers, or absent test methods are disqualifying red flags. Identical results across multiple batches are equally suspicious, since real manufacturing produces measurable lot-to-lot variation.
Key documentation red flags to screen for:
CoA shows only “Pass/Fail” with no numeric HPLC purity percentage
Batch number is absent or does not match shipping documentation
Testing date predates the manufacturing date or is missing entirely
No method reference for HPLC or mass spectrometry analysis
Acceptance criteria are listed without corresponding actual results
Facility address on the GMP certificate differs from the manufacturing site
Pro Tip: Request CoAs from at least three separate batches of the same peptide before approving a supplier. Comparing batch-to-batch variation in purity and impurity profiles reveals whether the supplier’s quality system is actually in control.
How does independent analytical testing verify peptide batch quality?
Independent third-party testing is the most reliable way to confirm that a supplier’s CoA reflects actual product quality. Third-party purity results should match the supplier CoA within ±1–2%, and deviations above 3% signal a quality system risk that warrants immediate investigation. This tolerance accounts for normal instrument variation between laboratories, not manufacturing inconsistency.
The standard testing panel for a new supplier qualification includes:
HPLC purity analysis to confirm the percentage of the target peptide relative to total peak area
Mass spectrometry (LC-MS) to verify molecular identity and detect truncated sequences or synthesis byproducts
Endotoxin testing (LAL method) for any peptide intended for injectable research applications
Residual solvent analysis to confirm that synthesis solvents such as DMF or NMP are below ICH Q3C limits
Moisture content by Karl Fischer titration, particularly relevant for lyophilized peptides where water activity affects stability
Independent analytical verification typically costs $150–$350 per compound. That cost is negligible relative to the risk of deploying a batch with undisclosed impurities in a research or clinical context.
Test Method What It Confirms Acceptance Benchmark HPLC purity Target peptide percentage ≥95% for research grade, ≥98% for API use LC-MS identity Molecular weight and sequence Matches theoretical MW ±0.5 Da Endotoxin (LAL) Bacterial contamination <1 EU/mg for research applications Residual solvents Synthesis solvent carryover Per ICH Q3C Class 2 limits Karl Fischer Moisture content Typically <5% for lyophilized peptides
When third-party results deviate from the supplier CoA by more than 2%, the correct response is a formal quality deviation investigation, not simply reordering. Request the supplier’s raw HPLC chromatogram and method validation data. If the supplier cannot produce these documents, that is itself a finding. For HPLC and LC-MS methodology in peptide analysis, the analytical approach directly affects how impurity profiles are interpreted.
Pro Tip: Send the same sample to two independent contract testing laboratories when qualifying a new supplier for the first time. Concordant results from two labs provide much stronger evidence than a single third-party result.
What aspects of a supplier’s quality management system indicate true GMP culture?
Documentation and test results reflect what a supplier produces. The quality management system (QMS) reflects how they respond when something goes wrong. Effective investigation and resolution of out-of-specification results reflect true GMP culture more than documentation alone. A supplier with a mature QMS will have written procedures for deviation classification, root cause analysis, and corrective and preventive action (CAPA) implementation.
Requesting a supplier’s deviation log summary or a redacted CAPA record is a legitimate part of supplier qualification. Suppliers who refuse to share any deviation history are not necessarily hiding failures, but the refusal itself is a data point. A supplier who can describe a specific OOS event, its root cause, and the corrective action taken demonstrates operational maturity. A supplier who claims to have no deviations is either very new or not tracking them.
Key QMS indicators to evaluate during a supplier audit or qualification review:
Written SOPs for synthesis, purification, lyophilization, and vialing are available and version-controlled
Deviation and OOS procedures define classification criteria and investigation timelines
CAPA records include effectiveness checks, not just corrective action descriptions
Quality agreements are offered as standard practice, not negotiated reluctantly
Technical staff can answer specific questions about Fmoc-SPPS synthesis steps, purification column selection, and endotoxin control measures
Supplier transparency about manufacturing and testing methods signals reliability. A supplier willing to share their synthesis platform, purification process, and method validation packages is operating with confidence in their own systems. Suppliers who deflect technical questions with marketing language are demonstrating the opposite.
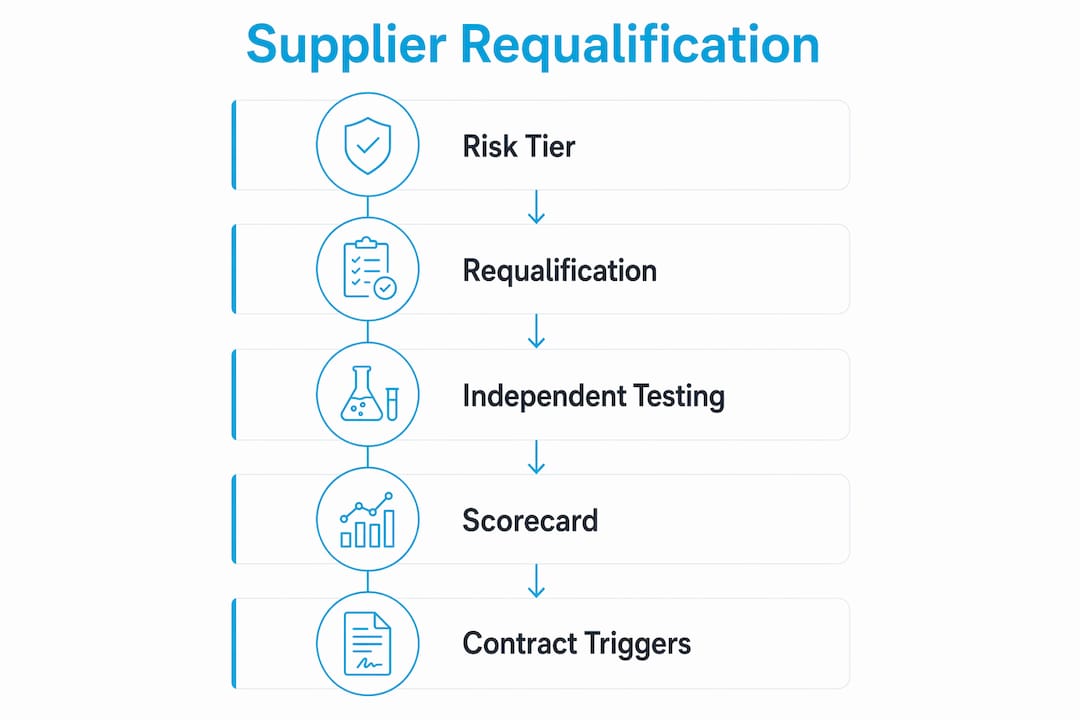
How to implement an ongoing supplier requalification program
A one-time qualification is not sufficient for suppliers providing peptides to ongoing research or commercial programs. FDA inspections check supplier qualification records under 21 CFR Part 211, and unqualified or inadequately monitored suppliers are a documented cause of recalls and warning letters. A formal requalification program converts supplier oversight from a reactive process into a managed risk control.
A structured ongoing monitoring program includes the following steps:
Classify suppliers by risk tier based on peptide criticality, regulatory status of the end product, and supplier audit history
Define qualification requirements per tier: documentation-only review for low-risk suppliers, full audit plus independent testing for high-risk or critical suppliers
Establish KPIs tracked per delivery: on-time delivery rate, CoA compliance rate, deviation frequency, and independent test concordance
Set requalification triggers: any critical deviation, a change in manufacturing site or ownership, or a lapse in regulatory certification
Schedule periodic requalification audits independent of triggers. Periodic requalification every 1–3 years is standard practice, with closer monitoring triggered by quality issues
Supplier Tier Risk Level Requalification Frequency Independent Testing Requirement Tier 1 (critical API) High Annual Every batch for first year, then quarterly Tier 2 (research grade) Medium Every 2 years First lot plus triggered testing Tier 3 (non-critical) Low Every 3 years First lot only
Supplier scorecards formalize this tracking. A scorecard records each delivery’s CoA compliance, any deviations reported, independent test results, and delivery performance. Reviewing scorecards quarterly allows procurement teams to identify deteriorating supplier performance before it becomes a supply chain failure. For a structured approach to procurement KPIs for peptide sourcing, tracking these metrics consistently is what separates a managed program from ad hoc oversight.
Pro Tip: Build requalification triggers directly into your supplier contracts. A clause requiring notification of any manufacturing site change, regulatory inspection, or ownership transfer gives procurement teams advance warning rather than post-hoc discovery.
Common pitfalls when evaluating peptide vendors for GMP compliance
The most common failure in peptide supplier evaluation is overreliance on supplier-provided documentation without independent verification. A supplier can produce a professionally formatted CoA that contains fabricated or selectively reported data. Without third-party testing on the first lot, there is no way to distinguish a compliant supplier from one who has learned to produce convincing paperwork.
Other frequent errors include:
Accepting ISO 9001 or ISO 13485 certification as equivalent to GMP compliance from a recognized regulatory authority
Failing to verify that the GMP certificate names the specific manufacturing site, not just the supplier’s commercial address
Ignoring small discrepancies in CoA data, such as purity values that are suspiciously consistent across batches
Skipping physical or remote audits because the supplier’s documentation appears complete
Treating a supplier’s evasive or defensive response to technical questions as a minor communication issue rather than a qualification finding
“A supplier who cannot explain their deviation handling process in specific terms has either never had a deviation or is not tracking them. Neither answer is acceptable for a GMP-compliant operation. The willingness to discuss failures openly is one of the most reliable indicators of a mature quality system.”
Handling supplier evasiveness requires a structured response. Document the specific questions asked and the responses received. If a supplier declines to share method validation data, a facility license, or a redacted CAPA record, record that refusal as a qualification finding. A pattern of evasiveness across multiple documentation requests is grounds for disqualification, regardless of how competitive the pricing is. For a detailed supplier vetting checklist, a structured document request process reduces the risk of missing critical gaps.
Key Takeaways
Effective peptide supplier qualification requires verified regulatory certificates, batch-specific CoA review, independent analytical testing, QMS evaluation, and a formal ongoing monitoring program to maintain supply chain integrity.
Point Details Verify certificates independently Confirm GMP certificates via FDA, EudraGMP, or NMPA databases, not supplier-provided copies alone. Require numeric CoA data Reject any CoA that shows only “Pass/Fail” without HPLC purity percentages and mass spectrometry confirmation. Test the first lot independently Third-party results must align with the supplier CoA within ±1–2% or trigger a formal quality investigation. Evaluate the QMS, not just paperwork Request deviation logs and CAPA records to assess whether the supplier’s quality system functions under real conditions. Requalify on a defined schedule Classify suppliers by risk tier and requalify every 1–3 years, with triggered reviews after any critical deviation or site change.
What I’ve learned from auditing peptide suppliers that most guides won’t tell you
The hardest part of supplier qualification is not the documentation review. It is the judgment call you make when a supplier’s paperwork is mostly correct but something feels off. I have reviewed CoAs from suppliers where the HPLC purity was reported to two decimal places with suspicious consistency across six consecutive batches. Real manufacturing does not produce that kind of uniformity. When I pushed for the raw chromatograms, two suppliers could not produce them. That alone was the answer.
The other lesson is that independent testing protects both parties. Procurement teams sometimes resist the cost of third-party testing on the grounds that the supplier already provided a CoA. But independent testing validates supplier CoAs and protects against lot-to-lot variability and shipping-related degradation. A peptide that left the facility at 97% purity can arrive at 91% after improper cold chain handling. The CoA will still say 97%. Only independent testing on the received lot catches that.
The most productive supplier relationships I have seen are ones where the QA team treated the initial qualification as a collaborative process rather than an interrogation. Sharing your acceptance criteria with the supplier upfront, explaining what your independent testing panel covers, and discussing their deviation history openly creates a working relationship where quality issues surface early rather than after a batch failure. That transparency does not compromise your standards. It raises the supplier’s.
— Sam Levin
PeptidesFromChina: GMP documentation and batch verification support
QA and procurement teams evaluating peptide suppliers need more than a product catalog. They need batch-specific CoAs, transparent analytical data, and a sourcing partner who can support the qualification process rather than complicate it.
PeptidesFromChina provides research-grade peptides with batch-specific CoAs that include HPLC purity data, mass spectrometry confirmation, and batch traceability documentation. The platform maintains direct relationships with established synthesis facilities, which means procurement teams can request method details and facility documentation rather than receiving deflection. Trial orders are available to support first-lot independent testing before full supplier approval. For teams building or updating a peptide vendor qualification program, PeptidesFromChina’s documentation standards are designed to meet the requirements of a formal supplier qualification review.
FAQ
What makes a GMP certificate valid for peptide supplier qualification?
A valid GMP certificate is issued by a recognized regulatory authority such as the FDA, EMA, or NMPA, names the specific manufacturing facility, and can be independently verified in the issuing authority’s official database. Self-issued certificates and ISO certifications do not satisfy GMP compliance requirements.
How often should peptide suppliers be requalified?
Periodic requalification every 1–3 years is standard, with the frequency determined by the supplier’s risk tier. Any critical quality deviation, manufacturing site change, or lapse in regulatory certification should trigger an unscheduled requalification regardless of the regular schedule.
What should a proper peptide CoA include?
A compliant CoA includes the peptide name and sequence, a unique batch number, testing date, HPLC purity results with method details, mass spectrometry data confirming molecular identity, and acceptance criteria listed alongside actual numeric results. A CoA showing only “Pass” without numeric data is not acceptable for GMP supplier qualification.
What is the acceptable purity deviation between a supplier CoA and independent testing?
Third-party purity results should align with the supplier CoA within ±1–2%. A deviation above 3% signals a quality system risk and requires a formal investigation, including a request for the supplier’s raw chromatographic data and method validation records.
How do you evaluate a peptide supplier’s quality management system?
Request written SOPs for synthesis and deviation handling, ask for a redacted CAPA record describing a specific OOS event and its resolution, and assess whether technical staff can answer specific questions about their synthesis platform and purification process. Suppliers who can describe real failures and their corrective actions demonstrate a functioning QMS.