How the Peptide Clinical Translation Process Works

Peptide clinical translation is the process of advancing a peptide drug candidate from laboratory discovery through clinical trials to regulatory approval and commercial use. The standard industry term is “clinical translation,” and it describes a decade-long sequence of scientific, manufacturing, and regulatory milestones that most peptide candidates fail to complete. Phase II trials alone involve 100–300 patients and carry high failure rates due to heterogeneous patient responses. Understanding each stage, and where projects typically collapse, is the difference between a productive development program and years of wasted resources.
How does the peptide clinical translation process work, stage by stage?
The clinical translation of peptides follows a defined sequence: discovery, preclinical evaluation, Phase I through Phase III clinical trials, regulatory submission, and post-marketing surveillance. Each stage has specific objectives, and failure at any point terminates the program.
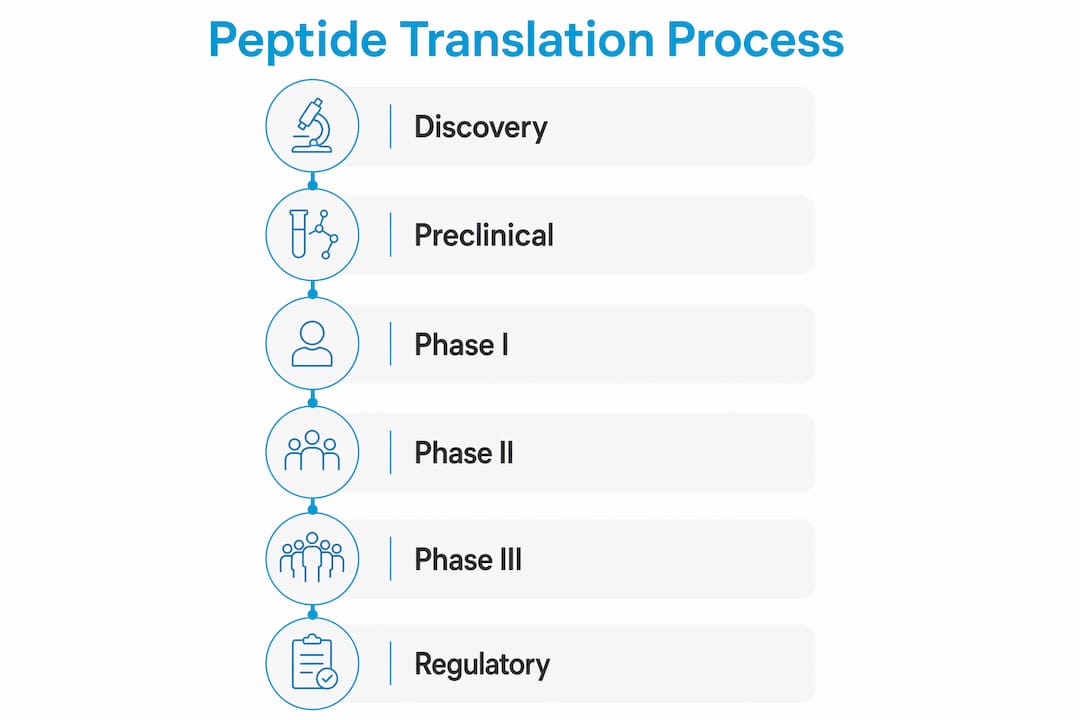
Discovery and early process development typically run 1–3 months for initial candidate identification. Researchers screen peptide sequences for target binding, selectivity, and preliminary stability. Process development follows over the next 4–6 months, covering synthesis route selection, purification method design, and early formulation work.

Preclinical evaluation uses both in vitro assays and in vivo animal models to assess toxicity, pharmacokinetics, and proof of concept. This stage generates the data package required for an Investigational New Drug (IND) application with the FDA. Preclinical work also informs dosing ranges for the first human studies.
Clinical phases proceed as follows:
Phase I tests safety and tolerability in a small group of healthy volunteers or patients. The primary goal is establishing a safe dose range and identifying adverse effects.
Phase II evaluates efficacy and refines dosing in 100–300 patients. This phase carries the highest attrition rate in the entire peptide drug development process, largely because computational models cannot fully predict heterogeneous patient responses.
Phase III runs confirmatory trials across larger, more diverse populations. Positive results support a New Drug Application (NDA) or Biologics License Application (BLA) submission.
Regulatory submission and review follows successful Phase III data. The FDA review process adds additional months to years before a decision. Post-marketing surveillance then monitors long-term safety in the general population. Peptide clinical translation usually takes over 10 years from first synthesis to market approval. That timeline reflects not just science, but the compounding effect of manufacturing delays, regulatory back-and-forth, and clinical recruitment challenges.
How does AI change the peptide drug development process?
Artificial intelligence accelerates several steps in the peptide drug development process, but it does not replace experimental validation. The distinction matters for anyone planning a realistic development timeline.
AI and machine learning tools currently contribute in three areas:
Sequence optimization: Models predict binding affinity and selectivity across large peptide libraries faster than wet-lab screening. In silico models can predict binding affinity values such as IC50 at approximately 1.4 nM, but those predictions require functional assay confirmation before advancing a candidate.
Stability and delivery prediction: Computational tools flag sequences prone to proteolysis or rapid renal clearance, two of the most common reasons peptides fail in vivo. This allows researchers to prioritize sequences with better pharmacokinetic profiles before committing synthesis resources.
Delivery system compatibility: AI models can screen formulation compatibility, predicting which delivery vehicles are likely to preserve peptide activity in physiological conditions.
The critical limitation is that computational tools cannot fully simulate in vivo complexity. Immune responses, protease activity in specific tissue environments, and off-target interactions require wet-lab validation regardless of how accurate the model appears in silico. AI accelerates candidate identification; it does not eliminate the need for rigorous experimental confirmation.
Pro Tip: Design delivery systems in parallel with potency optimization, not after. Retrofitting a delivery system to an already-optimized peptide sequence frequently compromises biological activity and adds months to the timeline.
The delivery-first integrated development approach treats molecular structure and delivery system as co-dependent variables from day one. GLP-1 analogs are the most cited example of this strategy working at scale. Programs that separate potency optimization from delivery design consistently encounter late-stage reformulation problems that extend timelines and inflate costs.
What manufacturing and technology transfer challenges affect peptide clinical translation?
Manufacturing preparation is one of the most underestimated sources of delay in steps in peptide translation. The transition from research-grade synthesis to GMP-compliant production involves documentation, method transfer validation, and process qualification at each new facility.
The standard timeline from discovery to Phase I GMP manufacturing readiness runs 25–30 months. That figure breaks down as follows: discovery (1–3 months), process development (4–6 months), technology transfers (7–18 months), and GMP manufacturing preparation (19–30 months). The technology transfer window is where most projects lose time.
When development moves across multiple standalone vendors, each transition requires re-establishing process knowledge. Standalone vendors cause 6–12 months of delay during development due to repeated route optimization and documentation challenges. Each new organization must re-validate methods, re-qualify equipment, and rebuild the process understanding that the previous team accumulated. That is not a bureaucratic problem. It is a structural one.
Integrated contract development and manufacturing organizations (CDMOs) address this by keeping discovery, process development, and GMP manufacturing under one operational roof. Integrated CDMOs reduce delays by preserving process continuity and eliminating redundant re-validation cycles. For complex peptides, including macrocyclic and stapled variants, this continuity directly affects whether scale-up meets GMP quality standards without costly reformulation.
Pro Tip: Request batch traceability records and method transfer documentation from any manufacturer before committing to a technology transfer. The absence of these records is the clearest signal that a vendor lacks the process discipline needed for clinical-grade production.
Batch traceability and independent quality control verification are non-negotiable for clinical-grade peptides. Researchers evaluating large-scale synthesis and sourcing options should treat the absence of documented batch records as a disqualifying factor, not a minor gap to address later.
How do regulatory frameworks affect clinical translation of peptides globally?
Regulatory classification determines the entire documentation burden for a peptide program. The classification is not uniform across jurisdictions, and misalignment between development strategy and regulatory pathway is one of the most expensive mistakes a team can make.
The FDA and India’s Central Drugs Standard Control Organization (CDSCO) illustrate the divergence clearly:
The FDA classifies peptides under 40 amino acids as small molecules, regulating them under the NDA pathway with Chemistry, Manufacturing, and Controls (CMC) documentation requirements aligned to small-molecule standards.
CDSCO treats the same peptides as biologics based on their mechanism of action and production process, triggering a different documentation framework and approval pathway.
This classification gap means a peptide approved under one framework may require entirely different CMC packages, stability data, and manufacturing standards to gain approval in the other jurisdiction.
Late-stage regulatory misclassification leads to costly delays and timelines extending by years. The cost of correcting a misclassified CMC package at Phase III is orders of magnitude higher than addressing it at IND submission.
Early engagement with regulatory consultants is the standard recommendation for programs targeting multiple markets simultaneously.
Researchers navigating FDA and CDSCO classification differences should map their target markets before finalizing CMC strategy. Building a documentation package that satisfies the more demanding jurisdiction first reduces rework when expanding globally. The lack of global regulatory harmonization for peptides makes this planning step mandatory, not optional.
Key Takeaways
Successful peptide clinical translation requires integrating scientific, manufacturing, and regulatory strategy from the earliest stages of development, not as sequential afterthoughts.
Point Details Translation takes over a decade The full process from discovery to approval typically exceeds 10 years, with Phase II carrying the highest attrition. Manufacturing prep is the hidden delay Technology transfers between standalone vendors add 6–12 months; integrated CDMOs preserve continuity and reduce this loss. AI accelerates but does not replace validation In silico tools predict binding affinity and stability, but wet-lab functional assays remain mandatory before advancing candidates. Regulatory classification drives documentation cost FDA and CDSCO classify the same peptides differently, making early regulatory strategy a financial necessity, not a formality. Delivery-first design improves success rates Optimizing molecular structure and delivery system simultaneously, as GLP-1 programs demonstrate, reduces late-stage reformulation failures.
Why the “design-first” mindset still costs programs years
The most persistent problem in peptide clinical translation is not scientific. It is organizational. Teams still design potent sequences first and treat delivery as a downstream problem. That sequence produces candidates that work beautifully in a binding assay and fail in vivo because no one addressed proteolytic stability or tissue penetration until Phase I data came back negative.
I have seen this pattern repeat across programs that had strong preclinical data and credible synthesis partners. The science was sound. The sequencing of decisions was not. The delivery-first approach described in the GLP-1 literature is not a novel concept at this point. It is the documented lesson from the programs that actually reached approval.
The second consistent failure mode is multi-vendor technology transfer without adequate documentation discipline. Teams underestimate how much process knowledge lives in the heads of the scientists who developed the synthesis route, not in the batch records. When that team hands off to a GMP facility, the receiving organization starts from a weaker position than anyone acknowledges. The 6–12 month delay figure is conservative for complex peptides. Macrocyclic and stapled sequences routinely extend that window further.
The regulatory piece is where I see the most avoidable damage. A team targeting both the US and Indian markets that builds its CMC package for the FDA pathway alone will face a complete documentation rebuild for CDSCO. That is not a translation problem. It is a planning failure. Regulatory consultants who specialize in peptide classification are not a luxury for programs with global ambitions. They are the cheapest investment in the entire development budget relative to the cost of late-stage corrections.
Realistic timeline planning and manufacturing transparency are not soft considerations. They are the variables that determine whether a program reaches Phase III or stalls in the gap between process development and GMP readiness.
— Sam Levin
Research-grade peptides for translation studies from PeptidesFromChina
Researchers working through early discovery and preclinical stages need peptide material that holds up to the scrutiny those stages demand. Batch inconsistency and undocumented purity at the research stage creates noise that compounds into larger problems when the program advances.
PeptidesFromChina supplies research-grade peptides with independent purity verification and batch traceability documentation included as standard. The catalog covers signaling peptides, GLP-1 analogs, longevity compounds, and custom synthesis options for programs with specific sequence requirements. Every batch ships with a Certificate of Analysis. Researchers who need verified material for translation studies can review the full catalog and sourcing documentation directly. Payment processing for international orders is supported through DavinciPay for nutraceuticals and related biotech procurement channels.
FAQ
How long does peptide clinical translation typically take?
Peptide clinical translation typically exceeds 10 years from initial discovery to regulatory approval, with manufacturing preparation alone requiring 25–30 months before Phase I can begin.
What causes the highest attrition rate in peptide clinical trials?
Phase II trials carry the highest failure rate due to heterogeneous patient responses and the inability of current computational models to fully predict in vivo biological complexity, including immune response and protease activity.
How does the FDA classify peptides for regulatory purposes?
The FDA classifies peptides under 40 amino acids as small molecules, requiring NDA submission with CMC documentation, while CDSCO in India treats the same peptides as biologics under a different approval framework.
What is a delivery-first approach in peptide development?
A delivery-first approach means optimizing the peptide’s delivery system simultaneously with its molecular potency, rather than retrofitting delivery after sequence optimization. GLP-1 analogs are the most documented example of this integrated strategy reaching commercial approval.
Why do technology transfers between vendors delay peptide programs?
Standalone vendors must re-validate synthesis methods and rebuild process knowledge at each handoff, adding 6–12 months to development timelines. Integrated CDMOs avoid this by maintaining continuity across discovery, process development, and GMP manufacturing.