Custom Peptide Sourcing for Product Development in 2026

Custom peptide sourcing in product development is the process of obtaining tailored peptides with defined sequence, purity, and production specifications to meet research or therapeutic innovation needs. For research scientists and product developers, the quality of that sourcing decision determines whether downstream assays, formulations, and regulatory submissions hold up under scrutiny. Suppliers like GenScript and Biosynth have built platforms around this demand, while technologies such as microwave-assisted synthesis have raised the bar for batch consistency and turnaround speed. Getting custom peptide sourcing and product development right from the start is not a procurement detail. It is a manufacturing readiness decision that shapes the entire development timeline.
What technical specifications must you define before ordering a custom peptide?
Defining technical parameters before placing an order is the single most effective way to prevent batch failures and reformulation cycles. Ambiguous specifications are the primary cause of peptide rejections at the analytical stage, not synthesis errors.
The core parameters to specify include:
Amino acid sequence and length. Longer sequences above 30 residues introduce coupling failure risk and require suppliers with validated long-chain synthesis protocols.
Post-translational and chemical modifications. Phosphorylation, biotinylation, PEGylation, disulfide bridging, and N-terminal acetylation each require distinct chemistry and add sourcing complexity.
Purity thresholds and analytical methods. Research-grade peptides typically require greater than 95% purity confirmed by reverse-phase HPLC and mass spectrometry. Clinical-stage material demands tighter specifications with full impurity profiling.
Batch size and scalability. Specify whether you need milligram quantities for assay development or gram-scale material for formulation studies. Batch size affects synthesis strategy and pricing.
Packaging and storage format. Lyophilized powder in sealed vials under inert gas is the standard for stability. Specify moisture barrier requirements and cold chain needs upfront.
Pro Tip: Request a certificate of analysis (COA) template from your supplier before ordering. If the COA does not include HPLC chromatograms, mass spec confirmation, and net peptide content by amino acid analysis, the documentation will not support regulatory submissions or reproducible research.
Sequence complexity drives cost and lead time more than any other variable. A 10-residue linear peptide with no modifications is a commodity synthesis. A 40-residue cyclic peptide with two disulfide bridges and a C-terminal amide is a project requiring process development. Treating these as equivalent sourcing decisions is a common and expensive mistake.
How to evaluate and select custom peptide synthesis suppliers
Supplier selection for custom peptide manufacturing is a qualification process, not a price comparison. The lowest quote frequently reflects compromises in raw material quality, analytical rigor, or batch traceability that only become visible after delivery.
The table below outlines the key evaluation criteria and what each reveals about supplier capability:
Evaluation criterion What it reveals GMP certification status Whether the facility operates under documented quality systems with audit trails Analytical control methods Depth of in-process and release testing, including HPLC, MS, and Karl Fischer moisture Batch traceability documentation Ability to trace raw amino acid lots through synthesis to final release Complex peptide track record Experience with long chains, cyclic structures, and non-standard modifications Small-batch to scale-up flexibility Whether the supplier can support early R&D through late-stage manufacturing Supply continuity planning Existence of secondary sourcing, safety stock, and contingency protocols
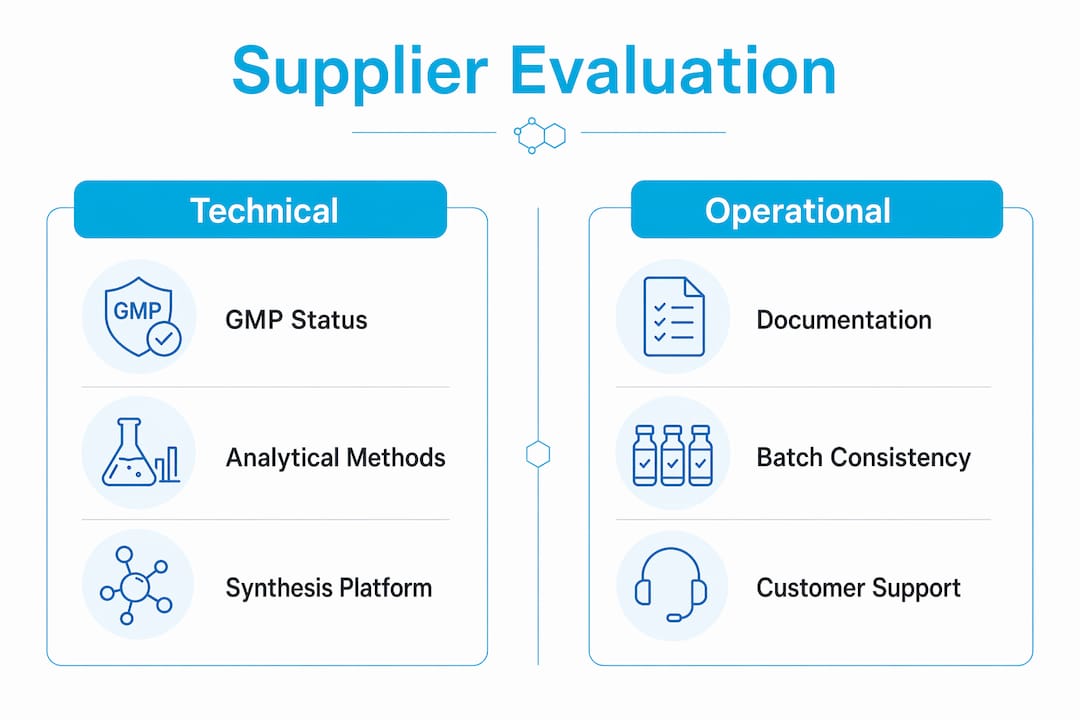
Supplier qualification should include review of GMP certification, technical capabilities, analytical control methods, and supply continuity plans as part of a risk-based quality-by-design approach. This is not a formality. Suppliers who cannot provide batch-specific analytical data on request are operating without the traceability infrastructure that reproducible research and regulatory submissions require.
Microwave-assisted synthesis platforms, used by providers like GenScript, deliver higher coupling efficiency and reduced racemization compared to conventional solid-phase synthesis. This matters when ordering peptides with sterically hindered residues or sequences prone to aggregation during chain assembly.

Pro Tip: Ask suppliers for a representative COA from a recent batch of a peptide with similar complexity to your target sequence. How they respond to that request tells you more about their quality culture than any marketing document.
The distinction between an API manufacturer and a reseller is critical in the Chinese peptide supply chain. Resellers purchase finished peptides from synthesis facilities and relabel them, often without independent analytical verification. Direct relationships with synthesis facilities provide batch-specific data, process visibility, and the ability to request reanalysis or reformulation when specifications are not met.
How do global regulatory requirements shape peptide sourcing decisions?
Regulatory requirements define the documentation, analytical standards, and manufacturing controls that custom peptide sourcing must satisfy before material can enter clinical or commercial development. Ignoring this dimension during early sourcing creates expensive remediation work later.
The FDA reviews peptide NDAs and BLAs expecting robust CMC data covering identity, purity, potency, and impurity profiling. CDER classifies synthetic peptides as drugs, which means the chemistry, manufacturing, and controls section of any submission must account for truncated sequences, epimerized residues, oxidized variants, and counter-ion content.
The EMA’s 2026 synthetic peptide guideline, finalized in 2024 and effective June 2026, mandates comprehensive impurity characterization and counter-ion control across EU member states. This guideline fills a regulatory gap that previously left synthetic peptide active substances without a dedicated quality framework in Europe. The practical implication is that suppliers providing material for EU-bound development programs must now demonstrate impurity profiling capabilities that many smaller facilities lack.
The table below summarizes key regulatory data requirements by agency:
Regulatory body Key CMC requirement Impurity focus FDA (CDER) Identity, purity, potency, full impurity profile Truncated sequences, oxidized variants, epimerization EMA Comprehensive impurity characterization, counter-ion control Synthetic process-related impurities, counter-ions Both Batch release documentation, stability data Degradation products under storage conditions
Outsourcing global regulatory strategy for peptides accelerates multi-market approvals and reduces compliance complexity by centralizing data package development and agency coordination. For organizations developing peptides across both U.S. and EU markets, a unified regulatory strategy compresses approval timelines and avoids the cost of sequential filings with inconsistent data packages.
Synthetic peptides and recombinant peptides follow different regulatory pathways. Synthetic material produced by solid-phase or liquid-phase synthesis falls under small-molecule drug frameworks at FDA, while recombinant peptides expressed in biological systems may require biologics licensing. Sourcing decisions made at the research stage determine which pathway applies, making early regulatory planning a sourcing constraint, not an afterthought.
What operational approaches support scaling from R&D to commercial manufacturing?
Scaling custom peptides from research quantities to commercial manufacturing requires a sourcing strategy built around supply continuity, process consistency, and supplier partnership depth. The transition from milligram to kilogram scale is where most development programs encounter their first serious supply chain failures.
The following sequence reflects industry best practice for managing that transition:
Qualify the supplier at the R&D stage. Supplier qualification performed during early research creates the documentation baseline needed for regulatory submissions. Waiting until Phase II to qualify a manufacturer adds months to the timeline.
Establish batch-to-batch consistency criteria early. Define acceptable ranges for purity, net peptide content, and residual solvent before scaling. These specifications become the release criteria that manufacturing must meet at every scale.
Plan for small-batch flexibility. Small-batch manufacturing enables dose flexibility, API conservation, and efficient early clinical phase transitions. CDMOs with small-batch capability allow dose-ranging studies without committing to large synthesis runs.
Engage a CDMO with integrated services. Contract Development and Manufacturing Organizations that offer formulation, fill-finish, and lyophilization under one roof reduce the handoff risk between synthesis and finished product manufacturing. Integrated drug product services covering preclinical through commercial manufacturing are the standard for late-stage peptide programs.
Build supply continuity into the contract. Specify safety stock requirements, lead time commitments, and secondary sourcing options in the supply agreement. A single-source supplier with no contingency plan is a development risk, not a partnership.
Align manufacturing strategy with the five S framework. TIDES manufacturing priorities for late-stage readiness center on scale, speed, supply security, sustainability, and strategic readiness. These five dimensions provide a practical checklist for evaluating whether a supplier can support a program from Phase I through commercial launch.
Process development at the synthesis level, including resin selection, coupling reagent optimization, and cleavage conditions, must be documented and transferred with the batch record when changing scale or site. Suppliers who treat process parameters as proprietary and refuse to share synthesis documentation create transfer risk that can delay regulatory submissions by a year or more.
Key takeaways
Reliable custom peptide sourcing requires defining technical specifications, qualifying suppliers on analytical rigor, aligning with regulatory requirements from the start, and building supply continuity into every stage of scale-up.
Point Details Define specifications upfront Sequence, modifications, purity thresholds, and packaging must be specified before ordering to prevent batch failures. Qualify suppliers on documentation Request batch-specific COAs with HPLC and MS data; distinguish API manufacturers from resellers. Regulatory requirements are sourcing constraints FDA CMC expectations and the EMA 2026 guideline define minimum analytical standards for supplier selection. Scale-up requires early supplier qualification Qualifying a manufacturer at the R&D stage creates the documentation baseline needed for regulatory submissions. Supply continuity must be contractual Safety stock, lead time commitments, and secondary sourcing options belong in the supply agreement, not in verbal assurances.
Why sourcing strategy is the decision most teams get wrong
The most consistent failure pattern in peptide product development is not a synthesis problem. It is a sourcing strategy problem that manifests as a synthesis problem six months into the program.
Teams routinely select suppliers based on price and turnaround time during the research phase, then discover at the IND-enabling stage that the supplier cannot produce the analytical documentation FDA expects. The remediation path requires requalifying a new supplier, generating fresh analytical data, and in some cases resynthesizing reference standards. That sequence adds cost and time that early supplier qualification would have avoided entirely.
The emerging complexity of peptide modifications is making this worse. Stapled peptides, cyclic peptides with non-natural amino acids, and peptide-drug conjugates each require synthesis capabilities and analytical methods that most catalog suppliers do not have. Sourcing these from a facility that has not validated the relevant chemistry is a reproducibility risk before it is a regulatory risk.
The cost-versus-quality calculation in peptide sourcing is also frequently misframed. The relevant comparison is not the cost of a high-quality batch versus a low-quality batch. It is the cost of a high-quality batch versus the cost of a failed batch plus the downstream consequences: lost time, repeated experiments, delayed submissions, and in clinical programs, potential patient safety implications.
PeptidesFromChina operates on the premise that supply chain transparency and independent batch verification are not premium features. They are the baseline for research-grade sourcing. That position is increasingly validated by the regulatory direction both FDA and EMA are moving in 2026.
— Sam Levin
How PeptidesFromChina supports your peptide sourcing workflow
PeptidesFromChina is a research-focused sourcing platform built around batch consistency, independent purity verification, and direct relationships with established synthesis facilities. The platform provides COA documentation with every order, including HPLC chromatograms and mass spec confirmation, so researchers and product developers can verify what they receive before it enters an experiment or formulation. For teams sourcing research-grade peptides across multiple programs, the catalog covers signaling peptides, longevity compounds, GLP-1 agonists, and laboratory materials with reproducible sourcing and supply continuity. Researchers working with specific compounds such as Epithalon can access verified material with full documentation, supporting both early-stage research and structured product development workflows.
FAQ
What is custom peptide sourcing in product development?
Custom peptide sourcing in product development is the process of procuring peptides with defined sequences, purity levels, and modifications from qualified synthesis suppliers to support research, formulation, or clinical programs. It differs from catalog sourcing in that specifications are set by the buyer rather than the supplier.
What purity level is required for research-grade custom peptides?
Research-grade custom peptides typically require greater than 95% purity confirmed by HPLC and mass spectrometry. Clinical-stage material requires tighter specifications with full impurity profiling to meet FDA and EMA CMC expectations.
How does the EMA 2026 peptide guideline affect sourcing decisions?
The EMA guideline, effective June 2026, mandates impurity characterization and counter-ion control for synthetic peptide active substances across EU markets. Suppliers providing material for EU development programs must demonstrate these analytical capabilities or the material cannot support regulatory submissions.
When should supplier qualification begin in a peptide development program?
Supplier qualification should begin at the R&D stage, not at the IND-enabling or clinical manufacturing stage. Early qualification creates the documentation baseline that regulatory submissions require and avoids costly requalification later in the program.
What is the difference between a peptide API manufacturer and a reseller?
An API manufacturer synthesizes peptides directly and can provide batch-specific process documentation, in-process analytical data, and synthesis records. A reseller purchases finished peptides from synthesis facilities and relabels them, typically without independent analytical verification or process visibility.